- Automated methods for reaction discovery
- Tools to analyze reaction networks
- Generalization of TSSCDS to study vdW complexes
- Energy transfer models in gas-surface collisions
- Rare-event acceleration
- Fitting PESs
- Vibrational angular momentum in MD simulations
1. Automated methods for reaction discovery
Over the last years we have been involved in the development of automated methods to discover complex reaction mechanisms. The methods are based on the use of molecular dynamics simulations, Heuristics and Graph Theory. The procedure has been applied to different systems and an open-source program is available:
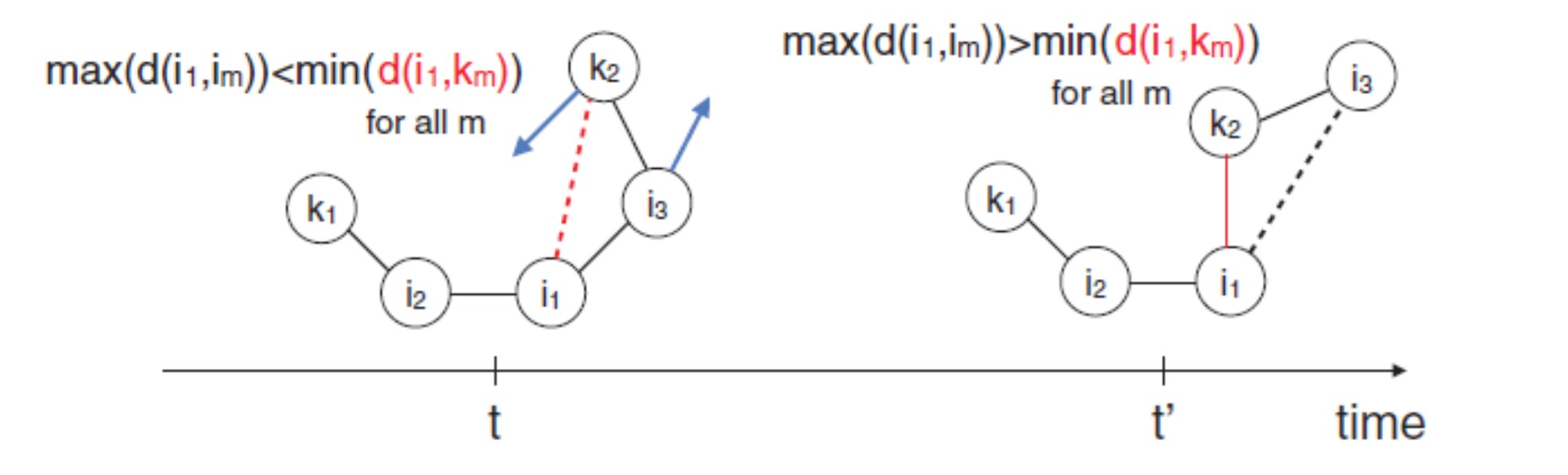
Example system showing the BBFS criterion, used in AutoMeKin to pindown the location of transition-state guess structures. Blue vectors correspond to the velocity vectors of the specific atom. Solid lines link neighboring atoms and the dashed lines indicate nonneighboring ones. At time step $\scriptstyle{t}$, the maximum distance between $\scriptstyle{i_1}$ and its neighbors $\scriptstyle{i_2}$, $\scriptstyle{i_3}$ is smaller than the minimum distance, $\scriptstyle{d}$, to non-neighbors $\scriptstyle{k}$; red dashed line. At time step $\scriptstyle{t’}$, the distance $\scriptstyle{d(i_1,k_2)}$ becomes smaller than the distance $\scriptstyle{d(i_1,i_3)}$, indicating the possibility of a TS in the vicinity.
$ \mathrm{max}\left[ d(i_1,i_m) \right] >\mathrm{min}\left[ d(i_1,k_m) \right] \quad \forall m $
The program and the underpinning methods are described in this publication. AutoMeKin mostly relies on the MD-based method developed under the name TSSCDS.
2. Tools to analyze reaction networks
The level of detail attained in the computational description of reaction mechanisms can be vastly improved through tools for automated chemical space exploration, particularly for systems of small to medium size. In this spirit, the new Python library has been designed to read and manipulate complex reaction networks, greatly simplifying their overall analysis. The package provides interactive dashboards featuring visualizations of the network, the three-dimensional molecular structures and vibrational normal modes of all chemical species, and the corresponding energy profiles for selected pathways.

Further details in the original publication. This is a collaborative work with Diego Garay-Ruiz, Moises Álvarez-Moreno, Carles Bo, from ICIQ.
3. Generalization of TSSCDS to study vdW complexes
We present a generalization of the transition state search using chemical dynamics simulations, TSSCDS, methodology, which allows the topographical characterization of intermolecular potential energy surfaces, IPES, for non-covalently bound complexes, vdW-TSSCDS. Starting from a single random input geometry, we show that vdW-TSSCDS is able to globally and automatically locate stationary points of an IPES, even in limiting cases such as extremely flat regions or nontrivial topologies, e.g., bifurcation points. The basic idea is the expression of the connectivity matrix in block structure, where diagonal blocks correspond to the isolated fragments and off-diagonal blocks provide the intermolecular connectivity. To this end, we introduce a new definition of bound or not, in a non-covalent sense, utilizing an extra set of van der Waals distances, which encompasses all kinds of non-covalent distances.
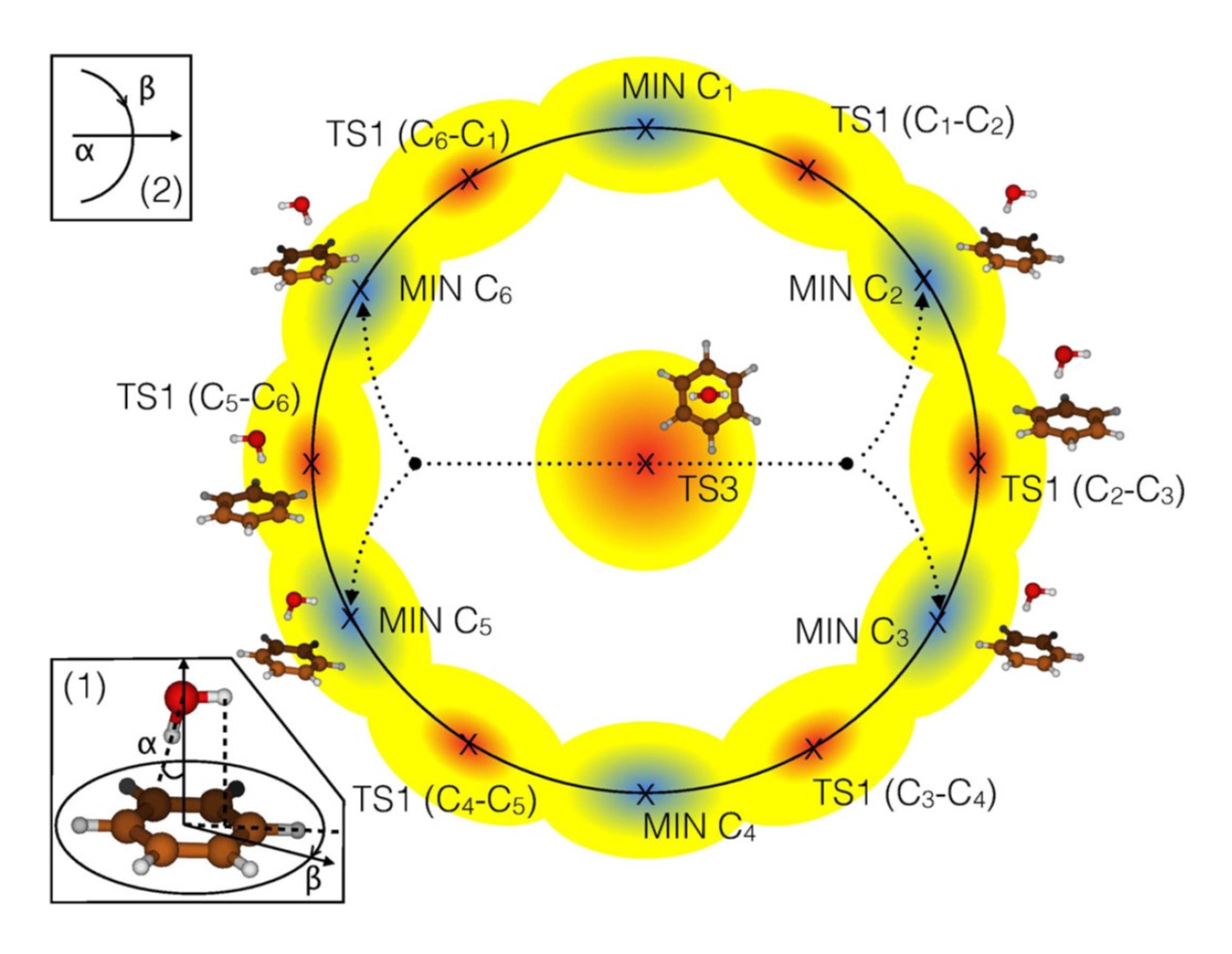
This work is a collaboration with Dani Peláez and this research group and this is the original work
4. Energy transfer models in gas-surface collisions
An energy transfer model has been developed for gas-surface collisions. The model relies on simple gas-phase scattering models. When energy transfer is analyzed in the limit of high incident energies, the following results are found:
-
The percent of energy transfer to vibration and rotation of light diatomic projectiles decreases as the projectile’s mass increases, while this transfer is almost independent of the mass for heavier projectiles.
-
For small projectiles, less than 10 atoms, transfer to vibration increases as a function of the projectile’s size.
-
However, for larger projectiles, the percent transfer to vibration is nearly constant, a result that can be attributed to a mass effect and also to the fact that only a reduced subset of “effective” vibrational dof is being activated in the collisions
The MD results can be very well fitted to an equation that accounts for the adiabatic, low-energy, and impulsive, high-energy, regimes:
$ \langle \Delta E \rangle=\langle \Delta E^{ad} \rangle+\langle \Delta E^{imp} \rangle=\langle \Delta E_0 \rangle+a\exp \left(\frac{-b}{\sqrt{E}}\right)+cd^2\mathrm{csch}^2\left(\frac{d}{\sqrt{E}}\right) $
with $\langle \Delta E_0 \rangle$, $a$, $b$, $c$ and $d$ being adjustable parameters.
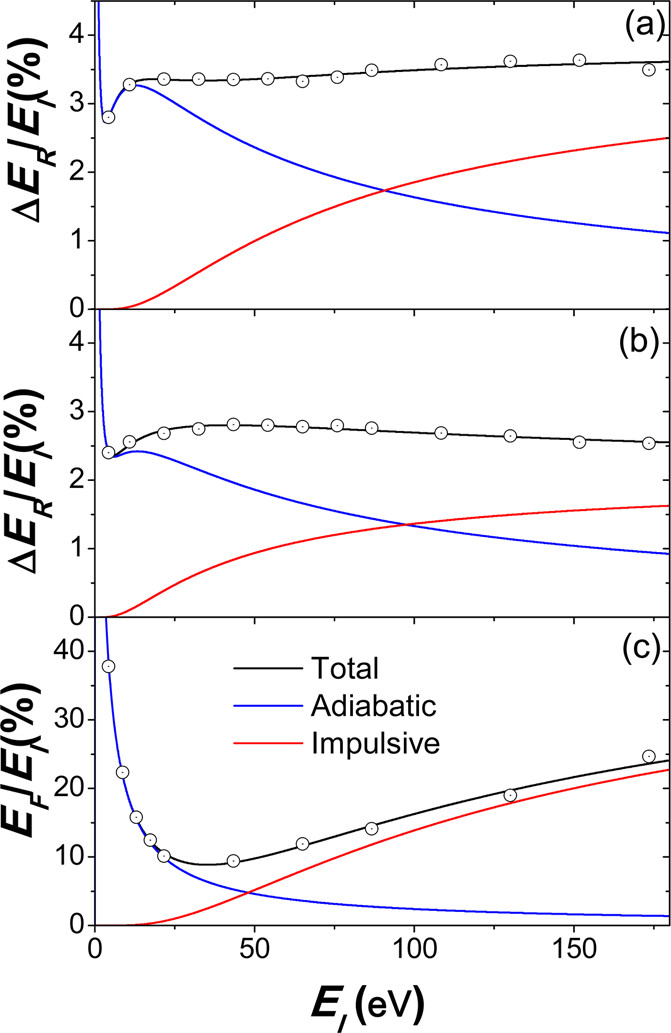
You can consult the full details in this publication
5. Rare-event acceleration
The approach is based on introducing constraints which lock trajectories in a region of the phase space close to the dividing surface with volume $\scriptstyle{\Gamma_1}$ , which separates reactants and products. This results in substantial, i.e., up to more than 2 orders of magnitude, speeding up of the trajectory simulation. Actual microcanonical rates $\scriptstyle{k(E)}$ are calculated by introducing a correction factor equal to the fraction of the phase volume which is allowed by the constraints. The constraints can be more complex than previously used boosting potentials. The approach has its origin in Intramolecular Dynamics Diffusion Theory, which shows that the majority of nonstatistical effects are localized near the transition state.
$ k(E)=k^{biased}(E)\frac{\Gamma_1}{\Gamma} $
The method was developed by Dmitry Shalashilin and myself and is described in this paper
6. Fitting PESs
We developed a software package based on a genetic algorithm that fits an analytic function to a given set of data points. The code, called GAFit, was also interfaced with the CHARMM and MOPAC programs in order to facilitate force field parameterizations and fittings of specific reaction parameters for semiempirical Hamiltonians. The present tool may be applied to a wide range of fitting problems, though it has been especially designed to significantly reduce the hard work involved in the development of potential energy surfaces for complex systems. For this purpose, it has been equipped with several programs to help the user in the preparation of the input files. We showcase the application of the computational tool to several chemical-relevant problems: force-field parameterization, with emphasis on nonbonded energy terms or intermolecular potentials, derivation of SRP for semiempirical Hamiltonians, and fittings of generic analytical functions.

This is a collaborative work with Saulo Vázquez and Roberto Rodríguez-Fernández, Francisco Pereira and Jorge Marques. Here is the publication
7. Vibrational angular momentum in MD simulations
Linear molecules with degenerate bending modes have states, which may be represented by the quantum numbers $\scriptstyle{N}$ and $\scriptstyle{L}$. The former gives the total energy for these modes and the latter identifies their vibrational angular momentum $\scriptstyle{j_z}$. In this work, the classical mechanical analog of the $\scriptstyle{N,L}$-quantum states is reviewed, and an algorithm is presented for selecting initial conditions for these states in MD simulations.
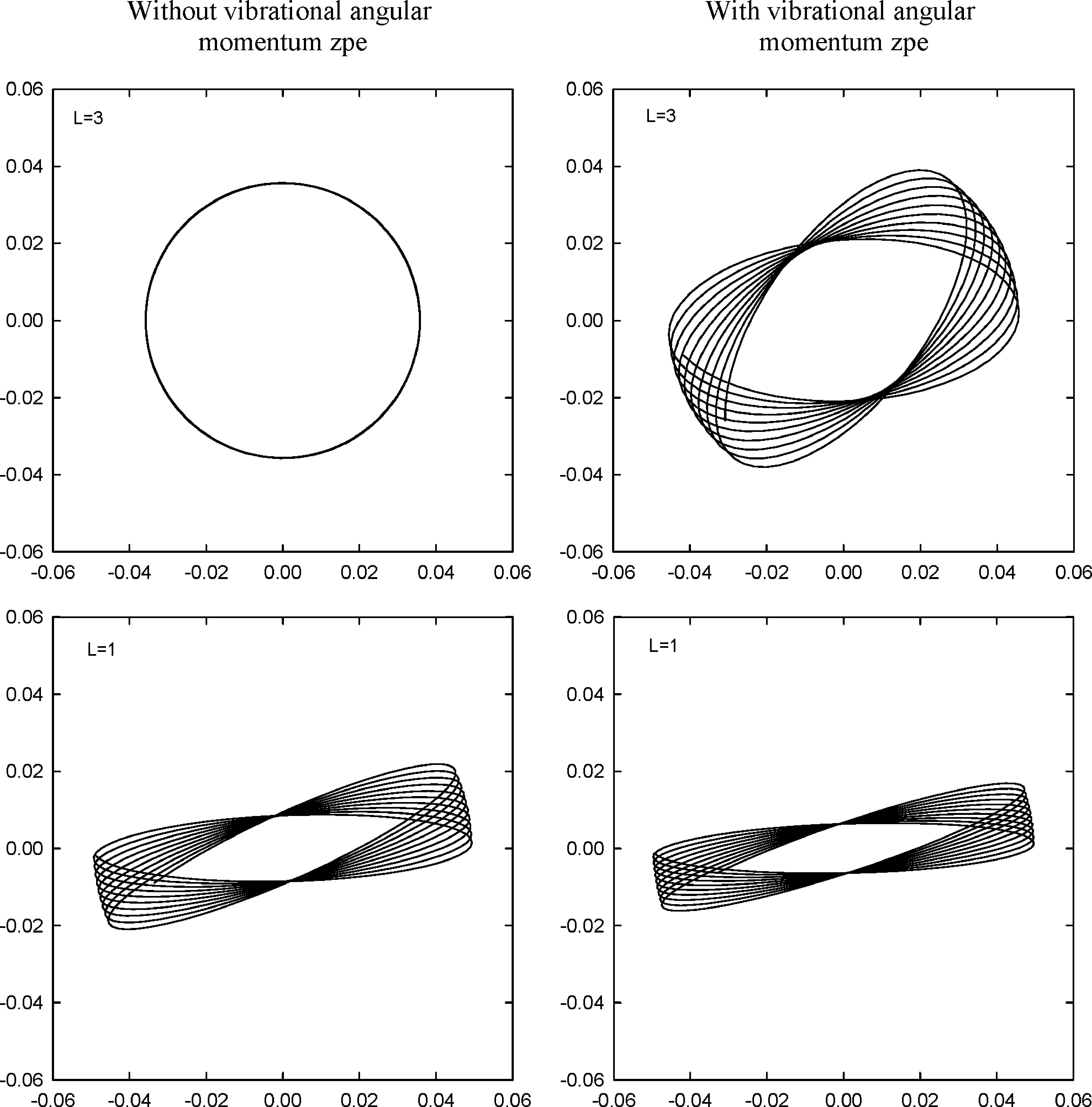
Plots of motion in the XY plane for an O-atom of CO2 prepared in different $\scriptstyle{L}$ states.
This is a collaborative work with Upakarasamy Lourderaj and Bill Hase$\scriptstyle{^\dagger}$. Here is the publication